Hauptinhalt
Molekulardynamik (MD) Simulation
Die Idee einer MD-Simulation ist, die klassischen Bewegungsgleichungen, d.h. Newton, zu numerisch zu integrieren, um daraus den zeitlichen Verlauf der Position eines Moleküls (Trajektorien) zu konstruieren. Dabei müssen alle beteiligten Atome berücksichtigt werden, insbesondere diejenigen der Umgebung. Meistens befinden sich die zu betrachtenden Teilchen auf einer Oberfläche oder in Lösung, wodurch die Anzahl der zu betrachtenden Teilchen in erster Näherung leider unendlich groß ist.
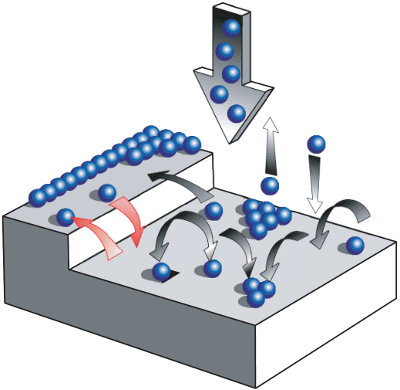
Abbildung 1: Am Filmwachstum sind zahlreiche Prozesse beteiligt (aus phys. stat. sol. (a) 201 (2004) Schreiber). Neben Bewegung über Stufenkanten und Nukleation ist vor allem die Diffusion auf der Substratoberfläche ausschlaggebend.
Da die Wechselwirkung mit der Umgebung i.d.R. auf einer deutlich schnelleren Zeitskala stattfindet, als die Bewegung des betrachteten Moleküls selbst, wird die Wirkung der Umgebung als ein auf das Molekül wirkendes Rauschen zusammengefasst. Das Ergebnis ist die Langevin-Gleichung. Dadurch ist beschrieben, wie das Molekül die thermische Energie der Umgebung in kinetische Energie umsetzt (Fluktuation) und wie es kinetische Energie an die Umgebung abgibt (Dissipation). Sowohl Fluktuation, als auch Dissipation hängen von Temperatur, Masse und Reibung ab, was über das Fluktuations-Dissipations-Theorem spezifiziert wird.
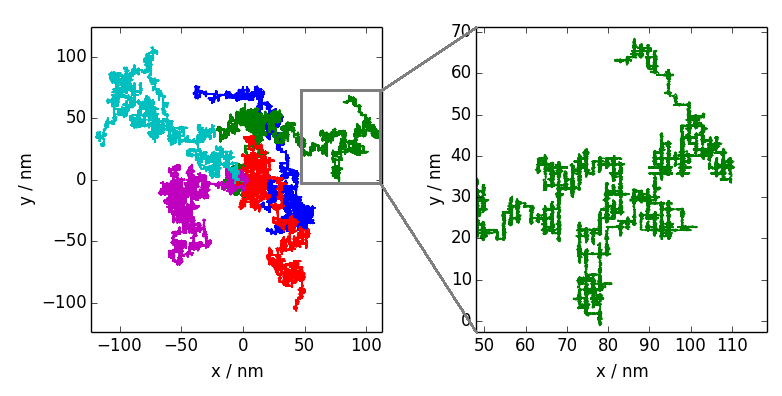
Abbildung 2: Dargestellt sind fünf Trajektorien eines Moleküls, welches sich über eine Oberfläche bewegt. Was auf den ersten Blick aussieht, wie gewöhnliche brownsche Bewegung, offenbart bei genauerer Betrachtung einer darunterliegende Struktur: Die Bewegung erfolgt hauptsächlich entlang eines rechteckigen Gitters.
Neben den erklärten thermisch fluktuierenden Kräften gibt es in der Langevin-Gleichung Wechselwirkungspotentiale zwischen diffundierenden Teilchen selbst und Oberfläche. Diese Wechselwirkung hängt stark von der Struktur des Systems ab und ist i.d.R. sehr aufwändig zu berechnen. Die Ansätze reichen von fundierten Vermutungen über Paarpotentialle (Molekularmechanik) bis zu quantenphysikalischen (DFT) Rechnungen.
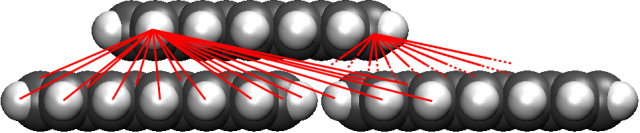
Abbildung 3a: Wechselwirkung von Pentacen mit einer Pentacenoberfläche. Das Wechselwirkungspotential wird durch paarweise Summation der Wechselwirkungen zwischen allen Kohlenstoff-Wasserstoff-Kombinationen berechnet.
Abbildung 3b: Pentacen diffundiert bei Raumtemperatur auf einer Pentacenobefläche. Die Pentacenmoleküle, welche die Oberfläche bilden, sind chemisch an eine Kupferoberfläche gebunden. Die in 5 Sekunden dargestellte Trajektorie entspricht einer Realzeit von 0,5 Nanosekunden. Die Animation ist also eine 1010-fache Verlangsamung.
Typische Fragestellungen sind Vergleiche mit theoretischen Vorhersagen oder Experimenten, welche sich aus den Trajektorien berechnen lassen.
Abbildung 4: Durch Auftragen des quadratischen Abstands über der Zeit erhält man einen linearen Zusammenhang mit der Diffusionskonstante als Proportionalitätsfaktor. Die obere Darstellung ist gemittelt aus den oben gezeigten fünf Trajektorien, wohingegen für untere Darstellung 300 Trajektorien berechnet und gemittelt wurden. Dadurch ist der Zusammenhang wesentlich deutlicher erkennbar.
Für aufwendige Simulationen steht in der AG ein den Bedürfnissen angepasster Hochleistungsrechner zur Verfügung.
Eine Publikation, in der MD in unserer Gruppe eingesetzt wurde:
- Coupling between diffusion and orientation of pentacene molecules on an organic surface.
Paul Rotter, Barbara A. J. Lechner, Antonia Morherr, David M. Chisnall, David J. Ward, Andrew P. Jardine, John Ellis, William Allison, Bruno Eckhardt, Gregor Witte
Nature Materials 15 (4), 397-400 (2016) • DOI: 10.1038/nmat4575
Full Text