Hauptinhalt
Massenbestimmung intakter Proteine und Proteinkomplexe

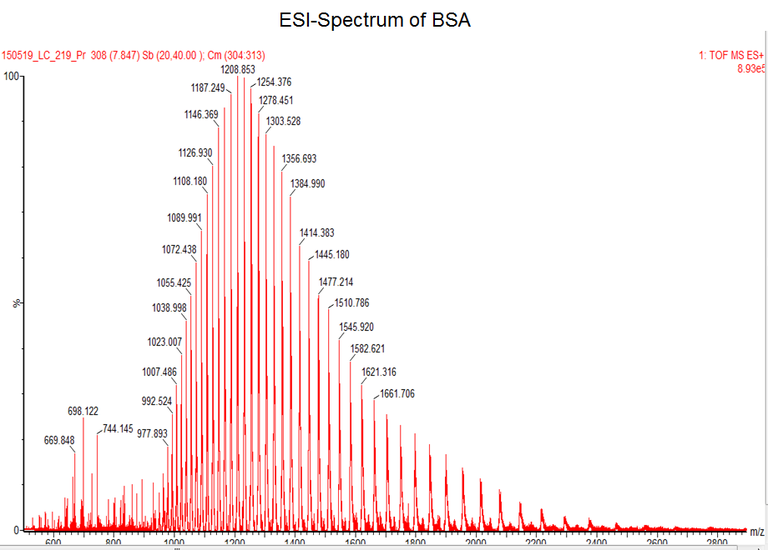
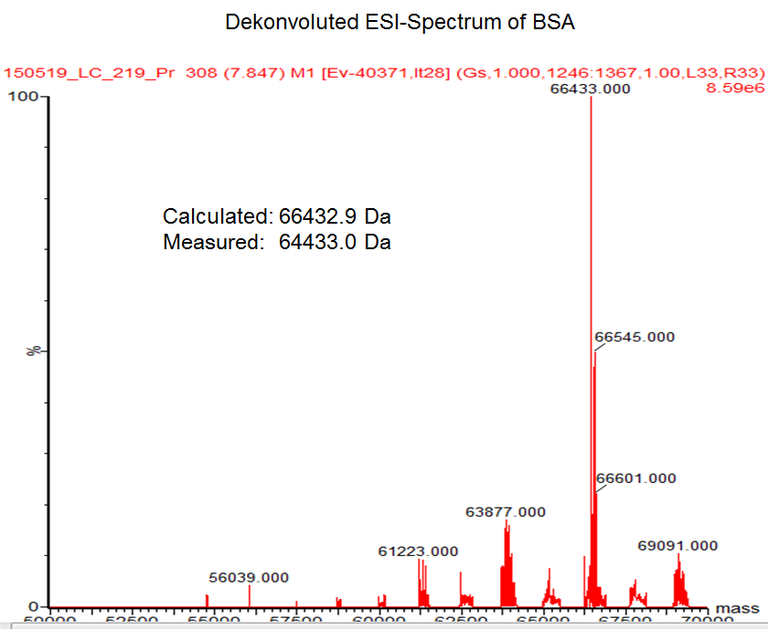
Take home message for mass determination of intact proteins:
Even masses of large intact proteins or protein complexes can be determined very accurate by ESI-MS
(standard accuracy ~ 10-20 ppm, which means +/- 1-2 Da at 100 kDA)
Bestimmung der intakten Proteinmasse, denaturierend (u.a. zur Qualitätskontrolle gereinigter Proteine oder zum Nachweis des Vorhandenseins von Modifikationen)
Bei dieser Methodik wird das Protein über eine RP-HPLC-Säule (in der Regel ein Monolith des Typs ProSwift RP 4H, oder auch eine klassische RP-C4) entsalzt und gleichzeitig denaturiert. Das entfaltete Protein wird mittels Electrospray-Ionisation (ESI) ionisiert und mit hochauflösender MS gemessen. Die Massengenauigkeit beträgt ca. 10ppm, was bei einem 100 kDa Protein einer Abweichung von max. 1 Da entspricht! Zu Unterscheiden ist die Massengenauigkeit aber von der Auflösung! Es ist mit dem verwendeten Gerät nicht möglich große Proteinspezies mit sehr ähnlichen Massen (wenige Da Unterschied) zu trennen! Die effektive Auflösung sollte im Bereich 5000-10.000 liegen. Im Einzelfall muss man es ggf. ausprobieren ob es funktioniert. Die Primärdaten enthalten eine Ladungsserie aus mehrfach geladenen Spezies. Als Faustregel gilt wie für alle mit ESI vermessenen Moleküle ca. 1 Ladung pro 1000 Da, so dass die Mehrheit der Signale im Bereich 1000-2000 m/z zu finden ist. Dadurch lassen sich praktisch alle Massenanalysatoren für die denaturierende Messung verwenden. Aus der gemessenen Ladungsserie erhält man dann durch den Prozess der Dekonvolution, der mit Hilfe bioinformatischer Software (in der Regel MaxEnt 1; Waters) erfolgt, die eigentliche Proteinmasse mit entsprechender Genauigkeit. Nicht kovalent gebundene Kofaktoren, etc. gehen bei der Denaturierung natürlich verloren - der Haupnachteil der Methode. Ein weiterer Nachteil ist, dass die Dekonvolution nicht ganz frei von Artefakten ist.
Die Methode eignet sich sehr gut als Qualitätskontrolle für gereinigte Proteine. Das Vorhandensein von posttranslationalen Modifikationen lässt sich ebenfalls schnell nachweisen. Zur zweifelsfreien Lokalisierung von Modifikationen ist aber ein proteolytischer Verdau notwendig. Bis ca. 30 kDa käme auch eine Top-Down-Sequenzierung des intakten Proteins mittels ETD als Alternative in Frage.
Probenbeschaffenheit: ca. 20 µL einer 5-50 µM Lösung, der Puffer kann frei gewählt werden.
Bestimmung der intakten Proteinmasse, nativ
Wie der Name bereits sagt wird die Probe hierbei in nativem, sprich gefalteten Zustand vermessen, was auch die Bestimmung nicht-kovalent gebundener Kofaktoren, Inhibitoren oder gar von Proteinkomplexen/Multimeren ermöglicht.
Da MS nicht kompatibel mit den meisten typischen salzhaltigen Puffern ist, muss das Protein aber vom Auftraggeber/der Auftraggeberin zunächst in einen MS-kompatiblen Puffer überführt werden. Geeignet ist eigentlich nur Ammoniumacetat oder -formiat bis zu einer Konz. von max. 100 mM. Keine weiteren "nicht-flüchtigen" Zusätze! Proteine, die unter diesen Bedingungen nicht stabil sind, sind nicht bzw. nur sehr schwer nativ zu vermessen. Im Einzelfall bitte nachfragen!
Theoretisch können Proteinkomplexe bis in den MDa-Bereich gemessen werden. Allerdings benötigt man dafür ein spezielles Setup. Da unser Gerät schwerpunktmäßig auch für HDX verwendet wird ist es eher für kleinere Moleküle konfiguriert (4K-Quadrupol, das bedeutet, dass er bis Massenbereich ca. 10.000 m/z ganz gut funktioniert). Für sehr große Komplexe wäre ein anderer Quadrupol nötig (8K oder gar 32K), der dann aber im Peptidbereich nicht optimal wäre. In der vorhandenen Konfiguration sollten native Messungen bis 200 kDa aber möglich sein, ggf. auch darüber hinaus. Das ist abhängig von den Proteineigenschaften und der daraus resultierenden Ladungsverteilung. Da zur Ladungsanlagerung (Protonen!) im Wesentlichen nämlich nur die Proteinoberfläche zur Verfügung steht ist deren Anzahl im Vergleich zur denaturierenden Messung natürlich deutlich geringer. Der Grundsatz 1 Ladung pro 1000 Da gilt hier nicht mehr! Je nach Proteingröße und Eigenschaften finden sich die Signale in einem erweiterten Massenbereich typischerweise oberhalb 2000 m/z, bei kleinen Proteinen teilweise auch darunter.
Die Messung erfolgt im manuellen Modus mit Spritzenpumpe und kann wegen des erweiterten Massenbereiches nur mit einem TOF-Massenspektrometer (in unserem Fall Synapt G2 Si von Waters) durchgeführt werden. Leider ist bei unserem Gerät im manuellen Modus softwarebedingt die Verwendung einer Lock-Masse für bessere Massengenauigkeit nicht möglich. Die zu erwartenden Abweichungen sind deshalb - und weil die Signale in einem höheren m/z-Bereich mit daraus resultierender niedrigerer Auflösung liegen, deutlich größer als bei der denaturierenden Messung. Eine sehr konservative Schätzung sind 1000 ppm (das entspricht 10 Da Abweichung bei einem 10 kDa Protein und 100 Da bei einem 100 kDa Protein; in der Praxis sollte die Abweichung in der Regel deutlich niedriger sein). Die Spritzenpumpe und die Kapillaren müssen gespült werden, so dass ein Verlust an Probe auftritt und ein deutlich größeres Volumen im Vergleich zur denaturierenden Messung notwendig ist.
Probenbeschaffenheit: min. 300-400 µL einer 5-50 µM Lösung in Ammoniumacetatpuffer ohne weitere "nicht-flüchtige" Zusätze wie z.B. NaCl