Hauptinhalt
Forschungsschwerpunkte
Das generelle Arbeitsgebiet der Arbeitgruppe ist die strukturbasierte Entwicklung neuer Wirkstoffe, wobei z. Zt. solche Substanzen im Vordergrund stehen, die potentiell als Anti-Infektiva einsetzbar sind. Die Entwicklung neuer Wirkstoffe orientiert sich dabei an bekannten Kristallstrukturen der Enzyme und erfolgt in enger Zusammenarbeit mit der Arbeitsgruppe von Prof. Klebe (Universität Marburg) und verschiedenen anderen Gruppen im In- und Ausland.
Aktuelle Arbeitsgebiete
Wirkstoffe gegen Schistosoma und andere humanpathogene Helminthen
Die Schistosomiasis stellt eine der „vernachlässigten tropischen Infektionserkrankungen (NTDs)“ von weltweiter Bedeutung dar und wird von Wurmparasiten der Gattung Schistosoma ausgelöst. Da es keinen Impfstoff für Schistosomiasis gibt und nur ein einziges, gegen alle Schistosomenarten wirksames Medikament (Praziquantel), haben die WHO und die G7-Staaten die Bekämpfung der NTDs einschließlich Schistosomiasis auf ihre Agenda gesetzt.
Biarylalkylcarbonsäuren
Ausgehend von dem Nachweis von Aldose-Reduktasen in Schistosoma mansoni haben wir einige Hemmstoffe der humanen Aldose-Reduktase, die aus früheren Untersuchungen zur Verfügung standen, gegen kultivierte Wurmpärchen getestet und dabei eine wirksame Verbindung identifiziert. Ausgehend von diesem „hit“ haben wir durch Strukturmodifikationen erste Struktur-Wirkungs-Beziehungen etabliert und Substanzen erhalten, die bei Konzentrationen bis 10 µM in vitro adulte Würmer töten und bisher nicht beobachtete Phänotypen erzeugen. Ausgehend von diesen Ergebnissen entwickeln wir die Substanzklasse der Biarylalkylcarbonsäuren weiter. Dazu werden umfangreiche Strukturvariationen durchgeführt und die Verbindungen in vitro getestet.
Dithiocarbamate
Ausgehend von dem anti-schistosomalen Effekt von Disulfiram (bis 25 µM; toxisch bei 25 µM) wurden in mehreren aufeinanderfolgenden Cyclen von Entwurf, Synthese und Testung gegen kultivierte Würmer mehr als 300 Dithiocarbamate der allgemeinen Formel 1 hergestellt und damit umfangreiche Struktur-Wirkungs-Beziehungen erarbeitet.
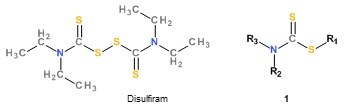
Die zur Zeit aktivste Verbindung führt zu multiplen Schädigungen des Wurms (Verlust innerer Strukturen, Tegumentschäden, massive Darmdilatationen. Bei 200 µM wurde keine Toxizität bei zwei humanen Zelllinien beobachtet.
Ziel der weiteren Arbeiten ist:
- Die Verbesserung der Aktivität und Erweiterung des Wirkungsbereichs auf andere Parasiten wie z.B. Fasciola, Echinococcus etc.
- Die Erhöhung der Wirkstoff-Ähnlichkeit der Moleküle
- Die Bestimmung und Verbesserung der ADTME-Parameter
Weitere Substanzklassen
Neben den beiden genannten Substanzklassen werden weitere Substanzklassen auf ihr Potential gegen Helminthen untersucht.
Die Arbeiten erfolgen in Zusammenarbeit mit Prof. Grevelding (Justus-Liebig Universität, Giessen) und Prof. Grünweller (Pharmazeutische Chemie, Philipps-Universität Marburg).
Entwicklung von Hemmstoffen der elF4A-spezifische RNA-Helikase als potenzielle Therapeutika gegen RNA-Viren insb. Ebola
Das EBOV besteht aus einem negativ orientierten Einzelstrang-RNA-Genom, das von einer viralen RNA-Polymerase in mRNAs mit einer 5´-Cap-Struktur transkribiert wird. Bei der Cap-abhängigen Translation wird der Translations-initiationskomplex eIF4F aufgebaut. Die RNA-Helikase eIF4A ist Bestandteil dieses Proteinkomplexes und für die Entwindung von RNA-Sekundärstrukturen verantwortlich.
Basierend auf der Raumstruktur eines wirksamen Naturstoffes wurden mehrere Strukturklassen kleiner Moleküle identifiziert, die als mögliche Hemmstoffe der Helikase elF4A geeignet erscheinen. In der Strukturklasse der Sulfonamide wurde bereits die angestrebten Aktivitäten gefunden. Diese Strukturklasse wird weiterentwickelt. Weitere Strukturklassen werden untersucht.
Die Arbeiten erfolgen in Zusammenarbeit mit Prof. Grünweller (Pharmazeutische Chemie, Philipps-Universität Marburg).
Entwicklung von Hemmstoffen der RNase P als potenzielle Therapeutika extrem pathogener Bakterien
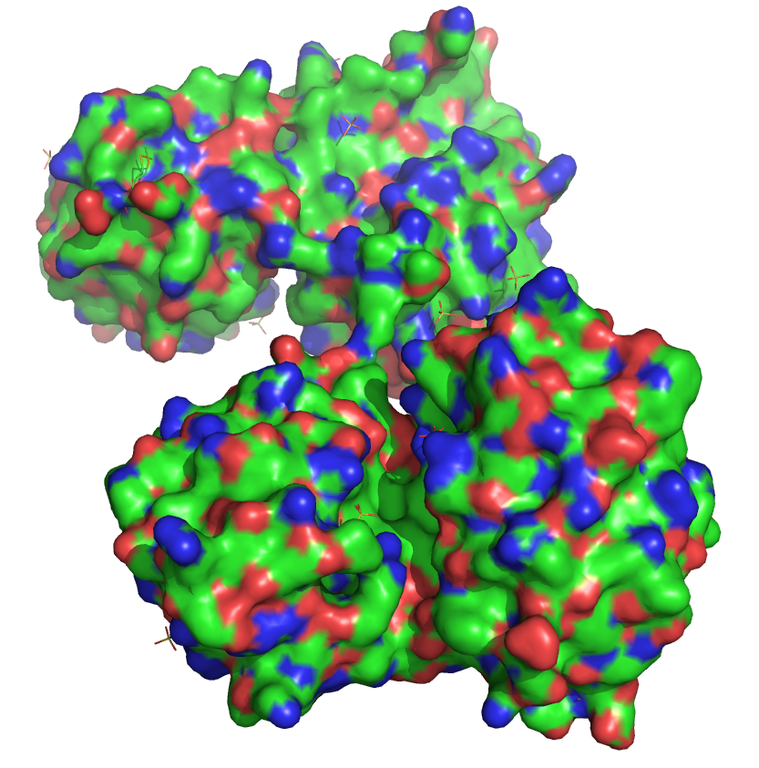
Die bakterielle Endoribonuklease P besteht aus einer katalytischen RNA und einem kleinen Protein und unterscheidet sich deutlich von den humanen RNase P-Enzymen. Ausgehend von der bekannten Raumstruktur der RNase P werden mögliche Liganden entworfen, synthetisiert und getestet. Die Bindetasche stellt aufgrund ihrer Form, Größe und Oberflächenstruktur eine besonders große Herausforderung für die Entwicklung möglicher Hemmstoffe dar.
Die Arbeiten erfolgen in Zusammenarbeit mit Prof. Hartmann und Prof. Kolb (beide: Pharmazeutische Chemie, Philipps-Universität Marburg).
frühere Arbeitsgebiete
Strukturbasierte Entwicklung von Farnesyltransferase-Inhibitoren
Die Farnesyltransferase katalysiert die kovalente Modifikation zahlreicher Proteine, die an der intrazellulären Signaltransduktion beteiligt sind. Die Substrate der Farnesyltransferase zeichnen sich durch eine C-terminale 4 Aminosäuren umfassende Konsensussequenz aus, die sogenannte CAAX-Box (C: Cystein; A: Aminosäure mit aliphatischer Seitenkette; X: Serin oder Methionin). Die Farnesyltransferase überträgt einen Farnesylrest von Farnesylpyrophosphat auf das Thiol der Cysteinseitenkette.
Entwicklung einer CAAX-peptidomimetischen Partialstruktur
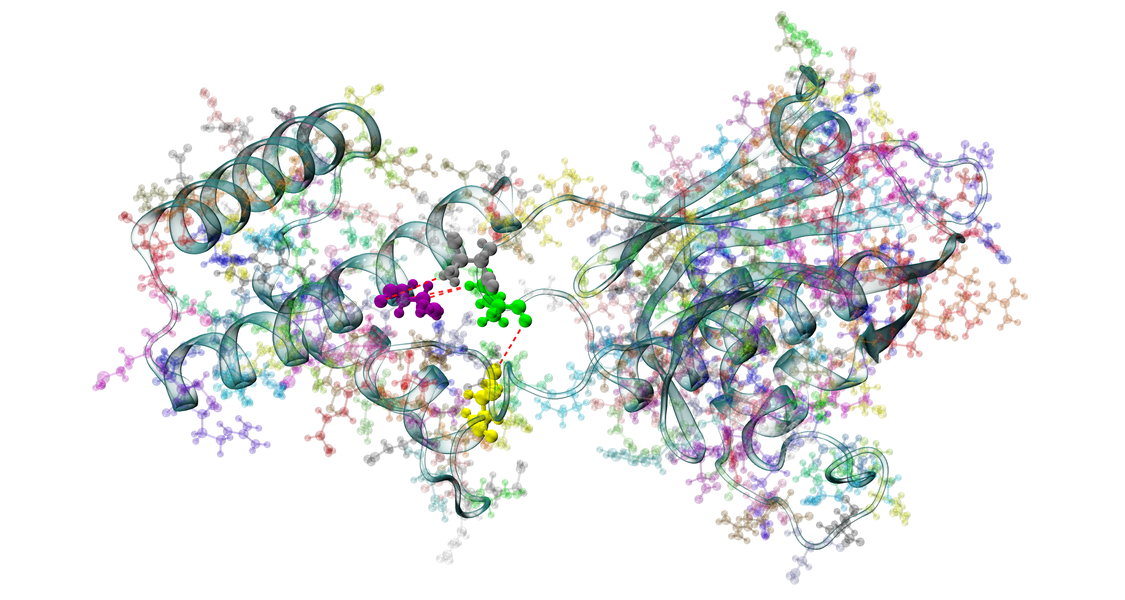
Zunächst haben wir eine eigene CAAX-peptidomimetische Partialstruktur entwickelt. Als wir mit diesen Arbeiten begonnen haben, war die Kristallstruktur der FTase noch nicht bekannt. Wir sind daher von dem natürlichen Substrat, dem CVIM-Tetrapeptid ausgegangen, haben dies in einzelne Partialstukturen zerlegt und diese dann in molekulare Eigenschaften übersetzt. Auf diesem Wege gelangten wir zu einem Pharmakophormodell, das wir anschließend in ein konkretes Molekül übersetzt haben. Das so erhaltene Benzophenon 1 hemmt die FTase mit einem IC50-Wert von 650 nM und qualifizierte sich damit als Leitstruktur einer neuen Klasse von FTase-Inhibitoren. Anschließende Strukturoptimierungen führten zu dem Derivat 2 mit annähernd 10fach verbesserter Wirksamkeit.
Anschließend haben wir dann Derivate unseres Benzophenon-Grundgerüsts entworfen, die in der Lage sind, mit einer Biarylstruktur spezifisch die oben gelb markierte Arylbindetasche zu besetzen. Die unter anderem so entstandene Substanz 3 weist eine gute FTase-Hemmung auf. Die Abb. zeigt die Verbindung 3 im aktiven Zentrum der Farnesyltransferase.
Durch weitere hier nicht gezeigte Modifikationen wurde die Aktivität dieser Verbindungen um eine weitere Größenordnung gesteigert und die Wasserlöslichkeit verbessert.
s. auch: M. Schlitzer Structure Based Design of Benzophenone-Based Non-Thiol Farnesyltransferase Inhibitors Curr. Pharm. Design 2002, 8, 1713-1722.
Farnesyltransferase-Inhibitoren als Anti-Malaria Wirkstoffe
Ursprünglich wurden Farnesyltransferase-Inhibitoren mit dem Ziel entwickelt, zu neuen Tumortherapeutika zu gelangen. Die Substanzen verschiedener Pharmafirmen befinden sich mittlerweile in fortgeschrittenen Stadien der klinischen Prüfung.
Farnesyltransferasen wurden jedoch auch in verschiedenen pathogenen Mikroorganismen wie z.B. in Plasmodium falciparum, dem Erreger der Malaria tropica, gefunden. Unterschiede in der Sequenz zwischen humaner und plasmoidaler FTase lassen die Entwicklung spezifischer Hemmstoffe möglich erscheinen.
Einige unserer Farnesyltransferase-Inhibitoren hemmen in vitro das Wachstum eines multiresistenten Plasmodienstamms in niedrigen nanomolaren Konzentrationen. Durch gezielte Modifikationen konnten zum ersten Mal Farnesyltransferaseinhibitoren erhalten werden, die in vivo (P. vinckei-infizierte Maus) gegen Malariaparasiten wirksam sind.
s. auch: J. Wiesner, K. Kettler, J. Sakowski, R. Ortmann, A. M. Katzin, E. A. Kimura, K. Silber, G. Klebe, H. Jomaa, M. Schlitzer Farnesyltransferase-Inhibitoren hemmen das Wachstum von Malariaerregern in vitro und in vivo Angewandte Chemie 2004, 116, 254 - 257; Angewandte Chemie Int. Ed. 2004, 43, 251 - 254.
Entwicklung von DOXP-Reduktoisomerase-Inhibitoren
Die Desoxy-D-Xylose-5-Phosphat-Reduktoisomerase (DXR) katalysiert einen Schlüsselschritt in der sog. Mevalonat-unabhängigen Isoprenoid-Synthese. Isopentenylpyrophosphat (IPP) ist der Baustein, von dem sich viele Isoprenoide ableiten. Die Biosynthese von IPP kann auf zwei alternativen Wegen erfolgen. Beim Mensch findet diese Synthese ausschließlich über den gut bekannten Mevalonat-Weg statt, während viele pathogene Mikroorganismen ausschließlich den DOXP-Weg nutzen. Enzyme dieses Stoffwechelweges stellen daher ausgezeichnete Zielstrukturen für die Entwicklung selektiver Wirkstoffe dar.
Die einzigen bekannten Inhibitoren der DXR sind Fosmidomycin und FR900098: Durch die Entwicklung verschiedener Prodrugs des Wirkstoffes FR900098 konnten wir die orale Resorption dieser Verbindung entscheidend verbessern.
s. auch: R. Ortmann, J. Wiesner, A. Reichenberg, D. Henschker, E. Beck, H. Jomaa, M. Schlitzer Acyloxyalkyl Ester Prodrugs Of FR900098 With Improved In Vivo Antimalarial Activity Bioorg. Med. Chem. Lett. 2003, 13, 2163 - 2166.
Aldose - Reduktase
Die Aldose-Reduktase katalysiert die insulin-unabhängige Reduktion von Glukose zu Sorbitol. Ein Zusammenhang zwischen dieser enzymatischen Umsetzung und der Entstehung diabetischer Spätschäden wurde nachgewiesen. Die Aldose-Reduktase stellt eine Herausforderung für die Wirkstoffentwicklung dar, da 1. ein Selektivitätsproblem zu der eng verwandten Aldehyd-Reduktase besteht, und 2. die Aldose-Reduktase in verschiedenen Konformationen existiert. In Zusammenarbeit mit der Arbeitsgruppe von Prof. Klebe (Uni Marburg) wurden Inhibitoren entwickelt, die spezifisch für einzelne Konformationen der Aldose-Reduktase sind.
Neuroprotektiva
Das Auslösen der intrinsischen Apoptose durch den Verlust des Membranpotenzials ist ein Schlüsselschritt neurodegenerativer Erkrankungen. Daher spielt die Regulation der neuronalen apoptotischen Prozesse eine wichtige Rolle. Auf der Grundlage des Inhibitors BI-6C9 wurden drei heterozyklische Grundstrukturen mit neuroprotektiver Wirkung gefunden. Diese verschiedenen Substanzklassen wurden derivatisiert und in Zusammenarbeit mit der Arbeitsgruppe Culmsee, FB 16, Marburg, auf neuroprotektive Wirkung untersucht.