Main Content
2008
SUR. SCI.: "Resolving the Depth Coordinate in Photoelectron Spectroscopy - Comparison of Excitation Energy Variation vs. Angular Resolved XPS for the Analysis of a Self-assembled Monolayer Model System"
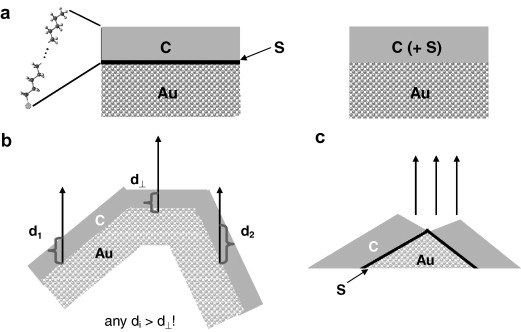
S. V. Merzlikin, N. N. Tolkachev, Th. Strunskus, G. Witte, Th. Glogowski, Ch. Wöll, W. Grünert
Surface Science 602 (3), 755-767 (2008), DOI: 10.1016/j.susc.2007.12.005
Methods for the quantitative XPS analysis of solids with concentration depth profiles in near-surface regions have been validated with reference samples consisting of self-assembled monolayers of n-octadecanethiol (“C18-SAM”) adsorbed on gold substrates of different surface roughness. In addition, they have been compared with respect to the effect of surface roughness on the results obtained. The roughness of the substrate surfaces has been studied by STM, AFM, and confocal laser scanning microscopy. Depth sensitivity was achieved by variation of the photoemission angle (ARXPS) and of the excitation energy (“ERXPS”). For the latter, a new data treatment algorithm has been developed in which intensity information acquired for different excitation energies is modelled with hypothetical depth distribution functions of the species studied, taking into account possible depth variations of the inelastic photoelectron mean free path. After identification of a suitable function type for the concentration depth profiles, the optimum profile parameters are determined. For a C18-SAM adsorbed on an atomically flat Au substrate this method allows to correctly identify the nature of the depth profile and to precisely determine the overlayer thickness and the (zero) gold concentration in it. While ARXPS yielded realistic results for the atomically flat Au substrate as well, it proved to be more affected by surface roughness than ERXPS. ARXPS may even completely fail to detect surface enrichment in carbon whereas ERXPS identified the organic surface layer though with exaggerated thickness. In ERXPS, the depth coordinate is extended by surface roughness, but the back-extrapolation to the external surface to describe the properties of the latter is not affected even for extremely rough surfaces. ERXPS offers, therefore, an attractive potential for the analysis of the outmost surface layer of real materials.PHYS. E: "Influence of contact metals on the performance and morphology of pentacene bottom-contact field-effect transistors"
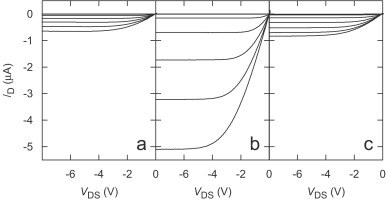
C. Bock, D. V. Pham, U. Kunze, D. Käfer, G. Witte, Ch. Wöll
Physica E 40 (6), 2107-2109 (2008), DOI: 10.1016/j.physe.2007.09.194
In pentacene-based bottom-contact field-effect transistors we study the influence of source and drain contact metals (gold, palladium, and platinum) on the morphology and transistor performance. Transistors prepared with Pd electrodes show a reduced contact and sheet resistance, a reduced activation energy, and a closer packed pentacene film in the channel region.PHYS. STAT. SOL.: "Molecular beam deposition and characterization of thin organic films on metals for applications in organic electronics"
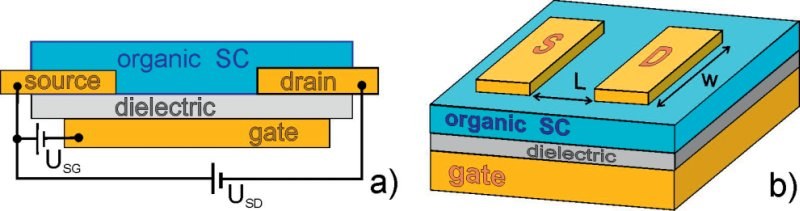
G. Witte and Ch. Wöll
physica status solidi (a) 205 (3), 497-510 (2008), DOI: 10.1002/pssa.200723433
The deposition of organic thin films on metal substrates using molecular beam deposition will be reviewed with a special emphasis on molecules which exhibit high charge carrier mobilities and are thus suited to be used as organic semiconductors (OSCs), namely pentacene, rubrene and perylene. Special emphasis will be on aspects of organic molecular beam deposition (OMBD) relevant for the device performance in organic field effect transistors (OFETs), in particular with regard to avoiding or minimizing structural defects at support/OSC interfaces. In addition, another aspect governing – and often limiting – charge injection at electrodes into an OSC, electronic level alignment at molecule/metal interfaces, will be discussed in the context of recent accurate ab-initio electronic structure calculations. Finally, we will present a novel experimental approach to determine charge transport properties of defect-free, nm-sized OSCs where extrinsic contributions to e.g. charge carrier mobilities can be strictly excluded, thus opening the way towards the determination of true intrinsic OSC properties.PHYS. REV. LET.: "Work function Changes Induced by Charged Adsorbates: Origin of the Polarity Asymmetry"
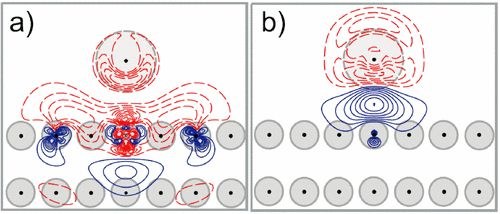
P. Bagus, D. Käfer, G. Witte, Ch. Wöll
Physical Review Letters 100 (12), 126101 (2008), DOI: 10.1103/PhysRevLett.100.126101
A theoretical analysis of charged adsorbates on a metal surface reveals a pronounced polarity asymmetry between electropositive and electronegative species, thus reproducing a well known but so far not properly understood experimental fact. For ionic adsorbates on metal surfaces, we analyze the several, often canceling, terms that contribute to the change of the interface dipole and, hence, to work-function changes, Δϕ. We demonstrate that for the prototypic case of I on Cu(111) the magnitudes and the signs of these terms can be understood on the basis of their physical and chemical origins. An important consequence of their cancellation is that negatively charged adsorbates can lead to a paradoxical Δϕ<0 rather than the expected Δϕ>0.ANG. CHEMIE: "Selenium as a Key Element for Highly Ordered Aromatic Self-Assembled Monolayers"
A. Bashir, D. Käfer, J. Müller, Ch. Wöll, A. Terfort, G. Witte
Angewandte Chemie Int. Ed. 47 (28), 5250-5252 (2008), DOI: 10.1002/anie.200800883
Less stress: Self-assembled monolayers of aromatic molecules become significantly better ordered, if selenium atoms instead of sulfur atoms are used as anchoring groups. Presumably as a result of a lowered corrugation for the Au–Se interaction, the molecules can adapt more easily to structures governed by the carbon backbone.ANG. CHEMIE: "Selen als Schlüsselkomponente für hochgeordnete aromatische selbstorganisierte Monoschichten"
A. Bashir, D. Käfer, J. Müller, Ch. Wöll, A. Terfort, G. Witte
Angewandte Chemie 120 (28), 5328-5331 (2008), DOI: 10.1002/ange.200800883
Stressabbau: Selbstorganisierte Monoschichten aromatischer Moleküle zeigen eine wesentlich bessere Ordnung, wenn als Ankergruppe Selen- statt Schwefelatome verwendet werden. Vermutlich führt eine geringere Korrugation der Au-Se-Wechselwirkung dazu, dass die Moleküle leichter Strukturen annehmen, die von den aromatischen Grundgerüsten vorgegeben werden.CRYS. GROW. DES.: "Packing of planar organic molecules: interplay of van-der-Waals and electrostatic interaction."
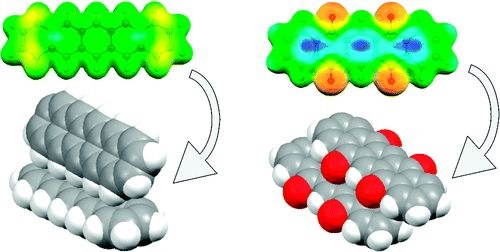
D. Käfer, M. El Helou, Ch. Gemel, G. Witte
Crystal Growth & Design 8 (8), 3053-3057 (2008), DOI: 10.1021/cg800195u
The molecular packing motifs occurring in the crystal structures of pentacene and its two oxo-derivatives (6,13-pentacenequinone and 5,7,12,14-pentacenetetrone) have been analyzed. Both oxygen containing species exhibit an almost coplanar stacking while pentacene adopts a face-on-edge herringbone packing. The different packing motifs are well explained by quantum chemical ab initio calculations of the electronic structure of the molecular entities exhibiting a pronounced charge localization at the oxygen atoms of both oxo-derivatives which causes an additional electrostatic O-π interaction favoring a planar stacking. Moreover, the polarizability of the π-system is reduced and the molecular quadrupole moment is altered, both resulting in a decrease of the lattice energy of the oxo-species as evidenced by the sublimation energy obtained for all three species from thermal desorption measurements. This emphasizes the importance of the balance between electrostatic and van der Waals interactions for the packing in molecular crystals.SUR. SCI.: "Reply to a comment of J. Zemek, Prague, regarding the paper ‘‘resolving the depth coordinate in photoelectron spectroscopy – comparison of excitation energy variation vs. angular-resolved XPS for the analysis of a self-assembled monolayer model system”"
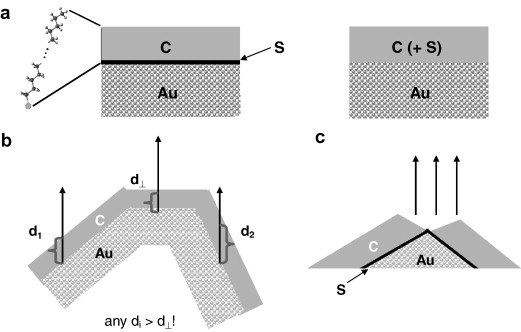
S. V. Merzlikin, N. N. Tolkachev, Th. Strunskus, G. Witte, T. Glogowski, C. Wöll, W. Grünert
Surface Science 602 (23), 3634-3635 (2008) • DOI: 10.1016/j.susc.2008.10.001
The molecular packing motifs occurring in the crystal structures of pentacene and its two oxo-derivatives (6,13-pentacenequinone and 5,7,12,14-pentacenetetrone) have been analyzed. Both oxygen containing species exhibit an almost coplanar stacking while pentacene adopts a face-on-edge herringbone packing. The different packing motifs are well explained by quantum chemical ab initio calculations of the electronic structure of the molecular entities exhibiting a pronounced charge localization at the oxygen atoms of both oxo-derivatives which causes an additional electrostatic O-π interaction favoring a planar stacking. Moreover, the polarizability of the π-system is reduced and the molecular quadrupole moment is altered, both resulting in a decrease of the lattice energy of the oxo-species as evidenced by the sublimation energy obtained for all three species from thermal desorption measurements. This emphasizes the importance of the balance between electrostatic and van der Waals interactions for the packing in molecular crystals.