Main Content
2023
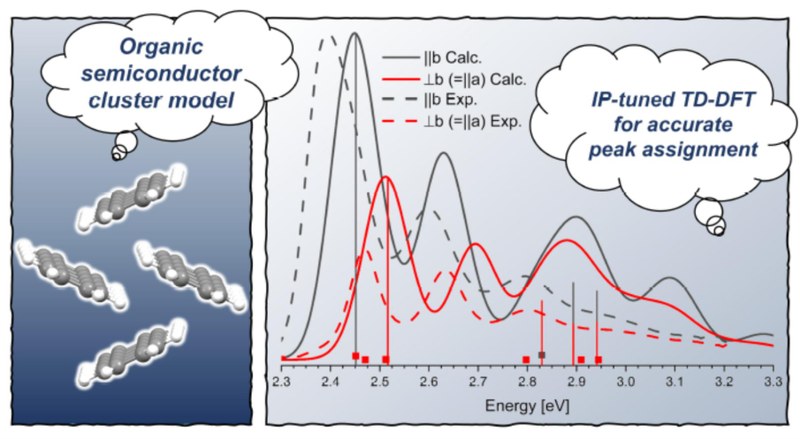
J. Chem. Theory Comp.: "Cluster-Based Approach Utilizing Optimally Tuned TD-DFT to Calculate Absorption Spectra of Organic Semiconductor Thin Films"
L. Craciunescu, M. Asbach, S. Wirsing, S. Hammer, F. Unger, K. Broch, F. Schreiber, G. Witte, A. Dreuw, P. Tegeder, F. Fantuzzi, and B. Engels
J. Chem. Theory Comp., 19, 24, 9369–9387 (2023), • DOI: 10.1021/acs.jctc.3c01107
The photophysics of organic semiconductor (OSC) thin films or crystals has garnered significant attention in recent years since a comprehensive theoretical understanding of the various processes occurring upon photoexcitation is crucial for assessing the efficiency of OSC materials. To date, research in this area has relied on methods using Frenkel–Holstein Hamiltonians, calculations of the GW-Bethe–Salpeter equation with periodic boundaries, or cluster-based approaches using quantum chemical methods, with each of the three approaches having distinct advantages and disadvantages. In this work, we introduce an optimally tuned, range-separated time-dependent density functional theory approach to accurately reproduce the total and polarization-resolved absorption spectra of pentacene, tetracene, and perylene thin films, all representative OSC materials. Our approach achieves excellent agreement with experimental data (mostly ≤0.1 eV) when combined with the utilization of clusters comprising multiple monomers and a standard polarizable continuum model to simulate the thin-film environment. Our protocol therefore addresses a major drawback of cluster-based approaches and makes them attractive tools for OSC investigations. Its key advantages include its independence from external, system-specific fitting parameters and its straightforward application with well-known quantum chemical program codes. It demonstrates how chemical intuition can help to reduce computational cost and still arrive at chemically meaningful and almost quantitative results.NPJ 2D Mater. and Appl., "Optical signatures of Förster-induced energy transfer in organic/TMD heterostructures"
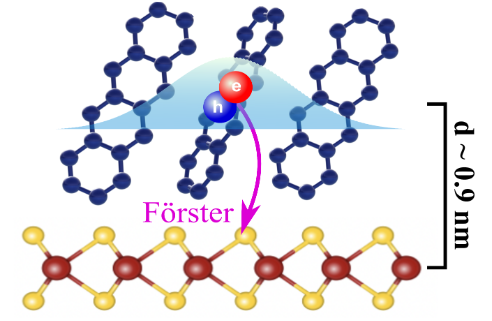
Joshua J. P. Thompson , Marina Gerhard, Gregor Witte and Ermin Malic
npj 2d materials and applications, 7, 69 (2023) • DOI: 10.1038/s41699-023-00430-z
Hybrid van der Waals heterostructures of organic semiconductors and transition metal dichalcogenides (TMDs) are promising candidates for various optoelectronic devices, such as solar cells and biosensors. Energy-transfer processes in these materials are crucial for the efficiency of such devices, yet they are poorly understood. In this work, we develop a fully microscopic theory describing the effect of the Förster interaction on exciton dynamics and optics in a WSe2/tetracene heterostack. We demonstrate that the differential absorption and time-resolved photoluminescence can be used to track the real-time evolution of excitons. We predict a strongly unidirectional energy transfer from the organic to the TMD layer. Furthermore, we explore the role temperature has in activating the Förster transfer and find a good agreement to previous experiments. Our results provide a blueprint to tune the light-harvesting efficiency through temperature, molecular orientation and interlayer separation in TMD/organic heterostructures.ELECTRON. STRUCT., "Excitons in organic materials: revisiting old concepts with new insights"
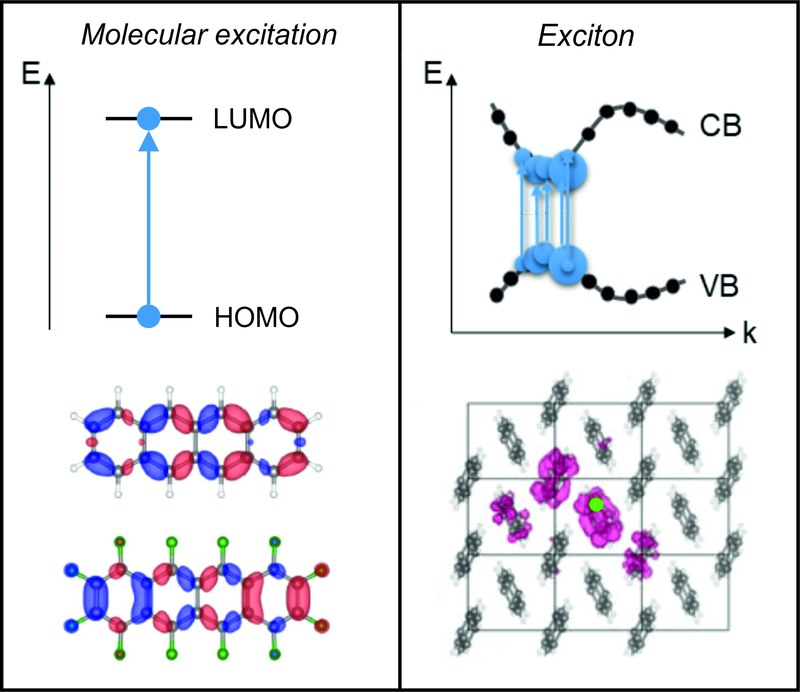
A. M. Valencia, D. Bischof, S. Anhäuser, M. Zeplichal, A. Terfort, G. Witte, C. Cocchi
Electron. Struct. 5, 033003 (2023), • DOI: 10.1088/2516-1075/acf2d4
The development of advanced experimental and theoretical methods for the characterization of excitations in materials enables revisiting established concepts that are sometimes misleadingly transferred from one field to another without the necessary disclaimers. This is precisely the situation that occurs for excitons in organic materials: different states of matter and peculiarities related to their structural arrangements and their environment may substantially alter the nature of the photo-induced excited states compared to inorganic semiconductors for which the concept of an exciton was originally developed. Adopting the examples of tetracene and perfluorotetracene, in this review, we analyze the nature of the excitations in the isolated compounds in solution, in the crystalline materials, and in melt. Using single crystals or films with large crystalline domains enables polarization-resolved optical absorption measurements, and thus the determination of the energy and polarization of different excitons. These experiments are complemented by state-of-the-art first-principles calculations based on density-functional theory and many-body perturbation theory. The employed methodologies offer unprecedented insight into the optical response of the systems, allowing us to clarify the single-particle character of the excitations in isolated molecules and the collective nature of the electron–hole pairs in the aggregated phases. Our results reveal that the turning point between these two scenarios is the quantum-mechanical interactions between the molecules: when their wave-function distributions and the Coulomb interactions among them are explicitly described in the adopted theoretical scheme, the excitonic character of the optical transitions can be captured. Semi-classical models accounting only for electrostatic couplings between the photo-activated molecules and their environment are unable to reproduce these effects. The outcomes of this work offer a deeper understanding of excitations in organic semiconductors from both theoretical and experimental perspectives.NAT. COMMUN., "Shape control in 2D molecular nanosheets by tuning anisotropic intermolecular interactions and assembly kinetics"
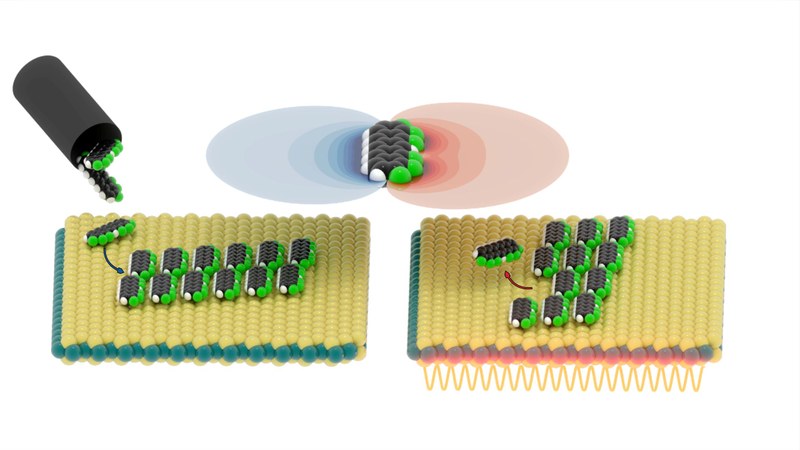
M. Dreher, P. M. Dombrowski, M. W. Tripp, N. Münster, U. Koert and G. Witte
Nat. Commun., 14, 1554 (2023), • DOI: 10.1038/s41467-023-37203-7
Since molecular materials often decompose upon exposure to radiation, lithographic patterning techniques established for inorganic materials are usually not applicable for the fabrication of organic nanostructures. Instead, molecular self-organisation must be utilised to achieve bottom-up growth of desired structures. Here, we demonstrate control over the mesoscopic shape of 2D molecular nanosheets without affecting their nanoscopic molecular packing motif, using molecules that do not form lateral covalent bonds. We show that anisotropic attractive Coulomb forces between partially fluorinated pentacenes lead to the growth of distinctly elongated nanosheets and that the direction of elongation differs between nanosheets that were grown and ones that were fabricated by partial desorption of a complete molecular monolayer. Using kinetic Monte Carlo simulations, we show that lateral intermolecular interactions alone are sufficient to rationalise the different kinetics of structure formation during nanosheet growth and desorption, without inclusion of interactions between the molecules and the supporting MoS2 substrate. By comparison of the behaviour of differently fluorinated molecules, experimentally and computationally, we can identify properties of molecules with regard to interactions and molecular packing motifs that are required for an effective utilisation of the observed effect.J. PHYS. CHEM. LETT., "Chemical Doping by Fluorination and Its Impact on All Energy Levels of π-Conjugated Systems
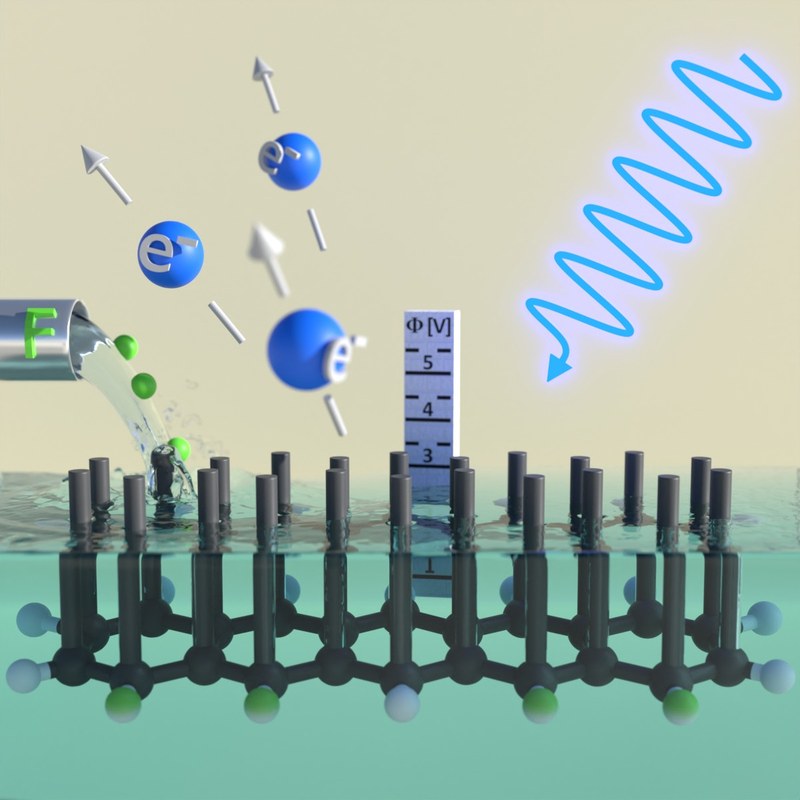
D. Bischof, Y. Radiev, M. W. Tripp, P. E. Hofmann, T. Geiger, H. F. Bettinger, U. Koert, and G. Witte
J. Phys. Chem. Lett., 14, 2551−2557 (2023), • DOI: 10.1021/acs.jpclett.3c00287
Halogenation of organic molecules causes chemical shifts of C1s core-level binding energies that are commonly used as fingerprints to identify chemical species. Here, we use synchrotron-based X-ray photoelectron spectroscopy and density functional theory calculations to unravel such chemical shifts by examining different partially fluorinated pentacene derivatives. Core-level shifts occur even for carbon atoms distant from the fluorination positions, yielding a continuous shift of about 1.8 eV with increasing degree of fluorination for pentacenes. Since also their LUMO energies shift markedly with the degree of fluorination of the acenes, core-level shifts result in a nearly constant excitation energy of the leading π* resonance as obtained in complementary recorded K-edge X-ray absorption spectra, hence demonstrating that local fluorination affects the entire π-system, including valence and core levels. Our results thus challenge the common picture of characteristic chemical core-level energies as fingerprint signatures of fluorinated π-conjugated molecules.