Main Content
Idiopathic achalasia
The aim of the international research network arc (achalasia risk consortium) is to elucidate the genetic- and cell-biological causes of achalasia. Molecular genetic analyses for the arc project are conducted at the Center for Human Genetics in Marburg. The arc website can be accessed under www.achalasie-konsortium.de.
Clinical features of idiopathic achalasia
Idiopathic achalasia is a disorder of esophageal motility. The disease is characterized by aperistalsis and a progressively defective relaxation of the lower esophageal sphincter (LES). As a result, ingested food can no longer be transported to the stomach. The disorder is attributable to a loss of inhibitory neurons in the myenteric plexus, which controls the muscle peristalsis of the esophagus and the opening of the LES. The precise cause of the neurodegeneration is not yet known. However, research has implicated autoimmune processes in individuals with a genetic predisposition (Gockel et al. (2010) Hum Genet). Idiopathic achalasia has a lifetime prevalence of 1: 10,000, and shows an equal sex incidence (Gockel et al. (2012) Dtsch Arztebl Int).
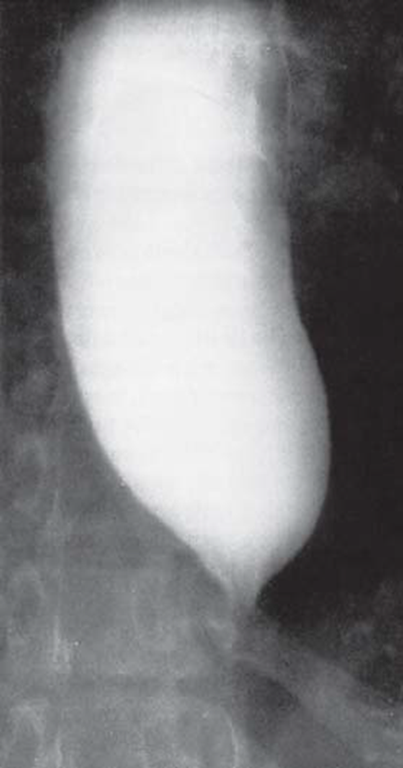
Clinically, idiopathic achalasia presents with difficulties in swallowing, regurgitation of food, retrosternal pain, and weight loss. Due to the restricted transport of food, the esophagus becomes massively dilated (megaesophagus) (see Figure 1).
If it enters the trachea, the pent-up food can lead to a broncho-pulmonary infection or aspiration pneumonia. Due to a chronic microaspiration of food, 50% of the patients show restricted lung function (Gockel et al. (2012) Dtsch Arztebl Int). Chemical and mechanical irritation of the mucous membrane secondary to food retention also leads to esophageal inflammation. As a result, 4-6% of patients develop squamous cell carcinoma (Gockel et al. (2012) Dtsch Arztebl Int). Available treatment options comprise surgical division of the LES (e.g., via a Heller cardiomyotomy), or pneumatic dilation of the esophagus.
Genetics of idiopathic achalasia
Idiopathic achalasia is a multifactorial disorder (Gockel et al. (2010) Hum Genet). To identify genetic risk variants for achalasia, we carried out the first systematic association analysis using a large European case-control collective (> 1,000 patients,> 2,200 controls) (Gockel et al. (2014) Nat Genet). The collective was used to test > 190,000 common genetic variants for association with the disease. The most strongly associated variant was rs28688207 (P = 1.73 x 10-19), which is located in the so-called HLA region on chromosome 6. The risk allele had a frequency of 9.9% in patients and 4.7% in controls (relative risk (RR) = 2.43). Functionally, the identified variant leads to the alternative splicing of an exon in the gene HLA-DQB1. At the protein level, the variant leads to an insertion of 8 amino acids in the cytoplasmic tail of HLA-DQβ1. We confirmed the association between achalasia and the insertion/genetic risk variant in an independent European case-control collective (> 400 patients,> 1,000 controls, 9.3% in patients, 3.8% in controls, P = 7, 09 x 10-09, RR = 2.59). We then tested whether independent achalasia risk variants were located in the HLA region of chromosome 6. This revealed a genome-wide significant association with achalasia for a genetic variant that leads - at the protein level - to an amino acid exchange at position 41 of HLA-DQα1. The amino acids lysine (K) and arginine (R) occur at this position, and it is the former which predisposes to disease (12.5% in patients, 7.1% in controls, P = 2.37 x 10-12, RR = 1, 86). We then tested whether other risk variants in the HLA region are located on chromosome 6. Here, we detected a third genome-wide significant association with achalasia. At the protein level, the respective genetic variant leads to an amino acid exchange at position 45 of HLA-DQβ1. The amino acids glutamic acid (E) and glycine (G) occur at this position. The former represents the achalasia risk allele (24.9% in patients, 22.4% in controls, 1.20 x 10-09, RR = 1.47).
At the protein level, HLA-DQβ and HLA-DQα form the heterodimer HLA-DQ complex. This is one of the HLA class II molecules, and is expressed on antigen-presenting cells (APC). HLA class II molecules present CD4 + T cell antigens, which are taken up extracellularly by phagocytosis or pinocytosis, and thus trigger an immune response within the organism. Our research data suggest that genetic variability in the HLA-DQ complex, and thus the immune response, is of key importance terms of the development of achalasia.
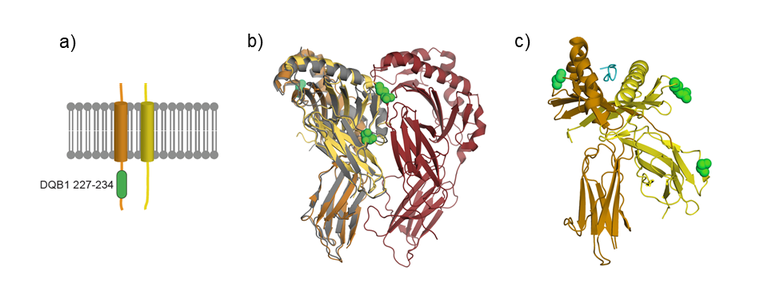
Finally, we projected the achalasia-associated variants into a 3D structure of the HLA complex at the protein level (see Figure 2). This should facilitate the generation of hypotheses concerning their cellular function.
In further studies, we investigated whether the most strongly associated achalasia- risk variant (rs28688207) - which is located in the HLA region on chromosome 6 and leads at the protein level to the 8-amino acid insertion in HLA-DQβ1 - shows an equal frequency across European subpopulations (Becker et al. (2016) Eur J Hum Genet). This revealed a geographical north-south gradient, with a higher frequency being observed in southern European populations (Italy, Spain) compared to central and northern European populations (Belgium, Germany, Netherlands, Poland, Sweden). We have since confirmed this north-south gradient using case control collectives from Greece, and from the Czech and Slovak Republics (Vackova et al. (2018) UEG Journal) (see Figure 3). The cause of this geographical north-south gradient, in which a higher frequency of the insertion is observed in southern Europe, is currently unknown. Evolutionary mechanisms, such as genetic drift or natural selection in the context of diverse environmental influences, are probably implicated. The latter study also showed that the 8-amino acid insertion in HLA-DQβ1 occurs more frequently in a specific subtype of idiopathic achalasia. Here, neurodegeneration of the inhibitory neurons in the plexus myentericus is particularly pronounced, as evaluated in accordance with the Chicago Classification (CC) and the results of high- Resolution Manometry (HRM).

To investigate the cell-biological causes of idiopathic achalasia in more detail, our group is currently working on two projects. In the first, we are investigating the pathophysiological consequences of the 8-amino acid insertion in HLA-DQβ1 at the cellular level. In the second, we are conducting the first extensive genome-wide association study (GWAS) of idiopathic achalasia using a large European case-control collective. Our aims are to identify individual genetic risk variants for idiopathic achalasia, and to determine the contribution of genetic factors that are shared with other (autoimmunological) diseases at the level of polygenic risk scores (PRSs).
Contact person
Prof. Dr. Johannes Schumacher
The arc website can be accessed under www.achalasie-konsortium.de.
Selected Publications
Vackova Z, Niebisch S, Triantafyllou T, Becker B, Hess T, Kreuser N, Kanoni S, Deloukas P, Schüller V, Heinrichs SKM, Thieme R, Nöthen MM, Knapp M, Spicak J, Gockel I, Schumacher J, Theodorou D, Martinek J. First genotype-phenotype study reveals HLA-DQB1 insertion heterogeneity in high-resolution manometry achalasia subtypes. United European Gastroenterology J. 2019 7:45–51.
Becker J, Niebisch S, Ricchiuto A, Schaich EJ, Lehmann G, Waltgenbach T, Schafft A, Hess T, Lenze F, Venerito M, Hüneburg R, Lingohr P, Matthaei H, Seewald S, Scheuermann U, Kreuser N, Veits L, Wouters MM, Gockel HR, Lang H, Vieth M, Müller M, Eckardt AJ, von Rahden BH, Knapp M, Boeckxstaens GE, Fimmers R, Nöthen MM, Schulz HG, Gockel I, Schumacher J. Comprehensive epidemiological and genotype-phenotype analyses in a large European sample with idiopathic achalasia. Eur J Gastroenterol Hepatol. 2016 28:689-95.
Becker J, Haas SL, Mokrowiecka A, Wasielica-Berger J, Ateeb Z, Bister J, Elbe P, Kowalski M, Gawron-Kiszka M, Majewski M, Mulak A, Janiak M, Wouters MM, Schwämmle T, Hess T, Veits L, Niebisch S, Santiago JL, de León AR, de la Serna JP, Urcelay E, Annese V, Latiano A, Fumagalli U, Rosati R, Laghi L, Cuomo R, Lenze F, Sarnelli G, Müller M, von Rahden BH, Wijmenga C, Lang H, Czene K, Hall P, de Bakker PI, Vieth M, Nöthen MM, Schulz HG, Adrych K, Gąsiorowska A, Paradowski L, Wallner G, Boeckxstaens GE, Gockel I, Hartleb M, Kostic S, Dziurkowska-Marek A, Lindblad M, Nilsson M, Knapp M, Thorell A, Marek T, Dąbrowski A, Małecka-Panas E, Schumacher J. The HLA-DQβ1 insertion is a strong achalasia risk factor and displays a geospatial north-south gradient among Europeans. Eur J Hum Genet. 2016 24:1228-31. doi: 10.1038/ejhg.2015.262.
Gockel I, Becker J, Wouters MM, Niebisch S, Gockel HR, Hess T, Ramonet D, Zimmermann J, Vigo AG, Trynka G, de León AR, de la Serna JP, Urcelay E, Kumar V, Franke L, Westra HJ, Drescher D, Kneist W, Marquardt JU, Galle PR, Mattheisen M, Annese V, Latiano A, Fumagalli U, Laghi L, Cuomo R, Sarnelli G, Müller M, Eckardt AJ, Tack J, Hoffmann P, Herms S, Mangold E, Heilmann S, Kiesslich R, von Rahden BH, Allescher HD, Schulz HG, Wijmenga C, Heneka MT, Lang H, Hopfner KP, Nöthen MM, Boeckxstaens GE, de Bakker PI, Knapp M, Schumacher J. Common variants in the HLA-DQ region confer susceptibility to idiopathic achalasia. Nat Genet. 2014 46:901-4. doi: 10.1038/ng.3029.
Wouters MM, Lambrechts D, Becker J, Cleynen I, Tack J, Vigo AG, Ruiz de León A, Urcelay E, Pérez de la Serna J, Rohof W, Annese V, Latiano A, Palmieri O, Mattheisen M, Mueller M, Lang H, Fumagalli U, Laghi L, Zaninotto G, Cuomo R, Sarnelli G, Nöthen MM, Vermeire S, Knapp M, Gockel I, Schumacher J, Boeckxstaens GE. Genetic variation in the lymphotoxin-α (LTA)/tumour necrosis factor-α (TNFα) locus as a risk factor for idiopathic achalasia. Gut. 2014 63:1401-9.
Gockel I, Müller M, Schumacher J. Achalasia--a disease of unknown cause that is often diagnosed too late. Dtsch Arztebl Int. 2012 109:209-14.
Gockel HR, Gockel I, Schimanski CC, Schier F, Schumacher J, Nöthen MM, Lang H, Müller M, Eckardt AJ, Eckardt VF. Etiopathological aspects of achalasia: lessons learned with Hirschsprung's disease. Dis Esophagus. 2012 25:566-72.
Gockel HR, Schumacher J, Gockel I, Lang H, Haaf T, Nöthen MM. Achalasia: will genetic studies provide insights? Hum Genet. 2010 128: 353-364.