Main Content
Stiewe Group
1. The p53 mutome
Mutations in the p53-encoding gene TP53 are observed in approximately half of all cancer patients, and germline mutations in human or mouse p53 invariably lead to cancer early in life. Different from other tumor suppressor genes, TP53 shows an unusual preference for missense mutations that result in the production of a full-length protein with single amino acid changes. Among the more than 80,000 cancer cases with somatic TP53 mutations reported in the Universal Mutation Database (UMD) TP53 database, ~73% carry missense mutations that affect more than 85% of all amino acid positions. Approximately 95% of these are located within exons 5-8 encoding the DNA binding domain that is crucial for p53’s function as a tumor suppressive transcription factor.
Figure: The TP53 gene locus and localization of somatic and germline missense mutations (figure from Thorsten Stiewe).
Literature:
Stiewe T, Haran TE (2018) How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resistance Updates 38: 27-43
1.1 A new class of p53 mutations – cooperativity mutations
p53 missense mutations in the DNA binding domain (DBD) are commonly subdivided into two classes: ‘DNA contact mutations’ affecting amino acids that directly contact DNA and ‘conformational mutations’ of residues that serve crucial functions in maintaining the 3D structure of the DBD. But not all mutations fit into the simple bipartite classification. DNA binding of p53 occurs in a tetrameric complex and requires stabilizing interactions between the DBDs of the four adjacent p53 monomers, which are established in the form of a double salt bridge by the residues E180 and R181. As these DBD interactions represent the structural basis for the cooperative nature of p53 DNA binding, mutations at these sites are referred to as ‘cooperativity mutations’ and affect world-wide an estimated number of 34,000 cancer patients per year. Our lab has therefore intensively characterized the consequences of cooperativity mutations for cancer development and cancer therapy responses using sophisticated cell culture and animal models. These studies have revealed remarkable phenotypic differences between cooperativity and classical p53 mutations and illustrate the still unresolved functional diversity of the complex p53 mutome.

Figure: Model of p53 DNA binding cooperativity mediated by a double salt bridge established by residues E180 and R181 (figure from Thorsten Stiewe).
Literature:
Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, Bretz AC, Sauer M, Charles JP, Vogiatzi F, Leich E, Samans B, Eilers M, Kisker C, Rosenwald A, Stiewe T (2010) DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 38: 356-68
Schlereth K, Charles JP, Bretz AC, Stiewe T (2010) Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle 9: 4068-76
Timofeev O, Schlereth K, Wanzel M, Braun A, Nieswandt B, Pagenstecher A, Rosenwald A, Elsasser HP, Stiewe T (2013) p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep 3: 1512-25
Timofeev O, Klimovich B, Schneikert J, Wanzel M, Pavlakis E, Noll J, Mutlu S, Elmshäuser S, Nist A, Mernberger M, Lamp B, Wenig U, Brobeil A, Gattenlöhner S, Köhler K, Stiewe T (2019) Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. In revision.
Funding:
DFG (SFB-TR17 project B2, STI 182/7-1), Deutsche Krebshilfe (#111250), Deutsche José Carreras Leukämie Stiftung (DJCLS R 13/08, DJCLS 09 R/2018)
1.2 Mutations switch p53 from a tumor suppressor into an oncogene
Missense mutations in some cases not only disrupt the tumor suppressor function of p53 (loss-of-function, LOF), but simultaneously endow the protein with novel oncogenic properties which actively promote tumor aggressiveness and limit patient survival. Oncogenic effectors of mutant p53 are therefore attractive therapeutic targets for a broad spectrum of cancer patients. We have recently identified ENTPD5 as a mutp53-induced target gene. ENTPD5 encodes for a UDPase enzyme, which is localized in the endoplasmic reticulum and promotes maturation of N-glycosylated membrane proteins such as growth factor receptors and integrins via the calnexin/calreticulin chaperones. Our experiments reveal ENTPD5 - just as mutant p53 itself - to be essential for migration, invasion and metastasis. This highlight ENTPD5 as a critical effector of mutant p53 and provide a conceptually novel link between the pro-metastatic activity of mutant p53 and glycoprotein folding in the endoplasmic reticulum.
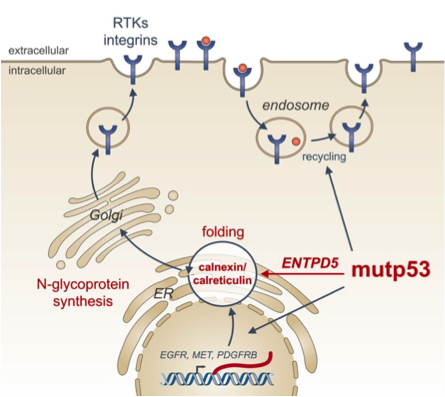
Figure: The endoplasmic reticulum UDPase ENTPD5 mediates mutant p53 effects on N-glycoprotein synthesis critical for metastatic tumor cell phenotypes (from Timofeev and Stiewe, Mol Cell Oncol 2017).
Literature:
Vogiatzi F, Brandt DT, Schneikert J, Fuchs J, Grikscheit K, Wanzel M, Pavlakis E, Charles JP, Timofeev O, Nist A, Mernberger M, Kantelhardt EJ, Siebolts U, Bartel F, Jacob R, Rath A, Moll R, Grosse R, Stiewe T (2016) Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc Natl Acad Sci U S A 113: E8433-E8442
Schneikert J, Stiewe T (2017) Pro-metastatic p53 mutants control folding of N-glycoproteins. Cell Cycle 16: 591-592
Timofeev O, Stiewe T (2017) p53 gain-of-function mutations promote metastasis via ENTPD5 upregulation and enhanced N-glycoprotein folding. Mol Cell Oncol 4: e1288678
Funding:
Deutsche Krebshilfe (#70112623)
1.3 p53: a druggable target for cancer therapy?
Cell fusion experiments in the 1960s showed that fusion of normal cells with tumor cells results in a non-malignant phenotype. This not only provided the experimental evidence for the existence of tumor suppressor genes, but also suggested restoration of tumor suppressors as a tumor therapy. Today, stimulated by the clinical success of oncogene-targeted drugs, there is a regained interest in therapeutic targeting of defective tumor suppressors like p53. However, fixing a broken tumor suppressor is technically more challenging than simply blocking an active oncogene. In fact, many putative ‘p53-reactivating compounds’ display considerable off-target activities, raising concerns whether the observed therapeutic responses are indeed caused by p53-reactivation. Our lab has therefore developed tools exploiting precise CRISPR-mediated TP53 gene editing to evaluate the p53-specificity of potential p53-reactiving compounds.
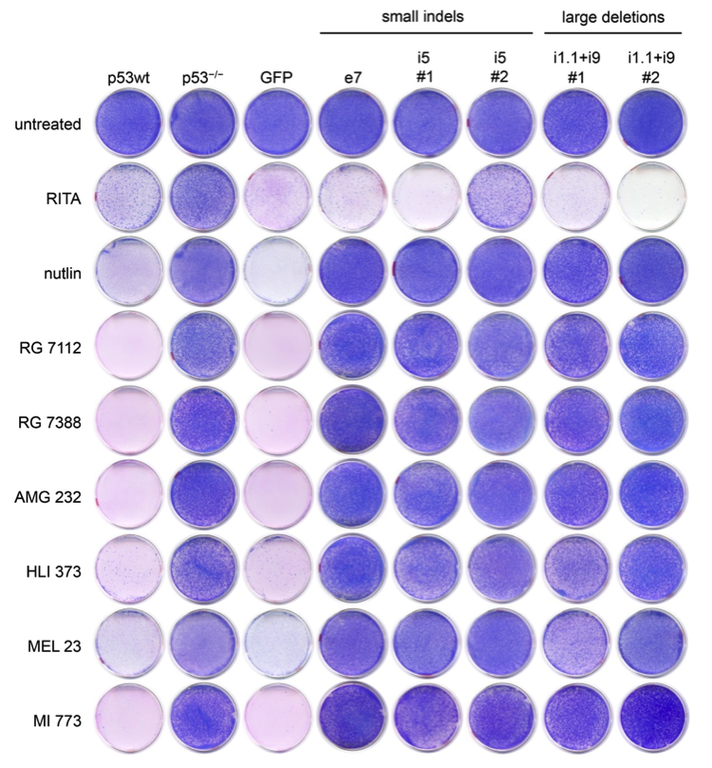
Figure: Colorectal cancer cells with CRISPR-engineered small indel or large deletion mutations become resistant to multiple Mdm2 inhibitors (nutlin, RG 7112, RG 7388, AMG 232, HLI 373, MEL 23, MI 773) but not to the proposed ‘p53-reactivating compound’ RITA. These results confirm the p53-specificity of multiple Mdm2 inhibitors except for RITA (figure from Michael Wanzel).
Literature:
Wanzel M, Vischedyk JB, Gittler MP, Gremke N, Seiz JR, Hefter M, Noack M, Savai R, Mernberger M, Charles JP, Schneikert J, Bretz AC, Nist A, Stiewe T (2016) CRISPR-Cas9-based target validation for p53-reactivating model compounds. Nat Chem Biol 12: 22-8
Funding:
DFG (KFO210 project 8; PI: Dr. Michael Wanzel)
2. Lung cancer
Lung cancer has the highest mortality rate among all types of cancer. Together with colleagues from the German Center for Lung Research (Deutsches Zentrum für Lungenforschung, DZL) we are investigating p53 family members in lung tumor development and cancer therapy responses. In addition, we are interested in p38MAPK functions in the crosstalk of lung cancer cells with their microenvironment.
2.1 p63 in squamous cell carcinoma
TP63, a member of the p53 gene family gene, encodes the DNp63 protein and is one of the most frequently amplified genes in squamous cell carcinomas (SCC) of the head and neck (HNSCC) and lungs (LUSC). Depleting DNp63 from SCCs resulted in reduced cell proliferation and sensitization to cisplatin-based chemotherapy. Using expression profiling we found DNp63 to induce the Fanconi anemia DNA repair pathway, thereby conferring relative resistance to DNA-crosslinking drugs such as cisplatin. Targeting DNp63 therefore would not only inhibit SCC proliferation but also sensitize tumors to chemotherapy.
Literature:
Bretz AC, Gittler MP, Charles JP, Gremke N, Eckhardt I, Mernberger M, Mandic R, Thomale J, Nist A, Wanzel M, Stiewe T (2016) DeltaNp63 activates the Fanconi anemia DNA repair pathway and limits the efficacy of cisplatin treatment in squamous cell carcinoma. Nucleic Acids Res 44: 3204-18
Funding:
DFG (KFO210 project 5)
2.2 Role of p73-directed chromatin alterations for neuroendocrine differentiation in small cell lung cancer
SCLC tumors frequently exhibit TP73 gene alterations of unclear relevance. We are investigating the role of p73-directed chromatin changes for neuroendocrine differentiation and the development of SCLC.
Funding:
ERC (p73CANCER), DFG (SFB-TRR81 project A10)