08.09.2022 IgG against the membrane-proximal portion of the desmoglein 3 ectodomain induces loss of keratinocyte adhesion, a hallmark in pemphigus vulgaris
Published on September 8th, 2022 in the Journal of Investigative Dermatology
Christoph Hudemann, Yvonne Exner, Robert Pollmann, Karina Schneider, Anna Zakrzewicz, Simon Feldhoff, Thomas Schmidt, Volker Spindler, David Rafei-Shamsabadi, Frauke Völlner, Jens Waschke, Ritva Tikkanen, Michael Hertl, Rüdiger Eming
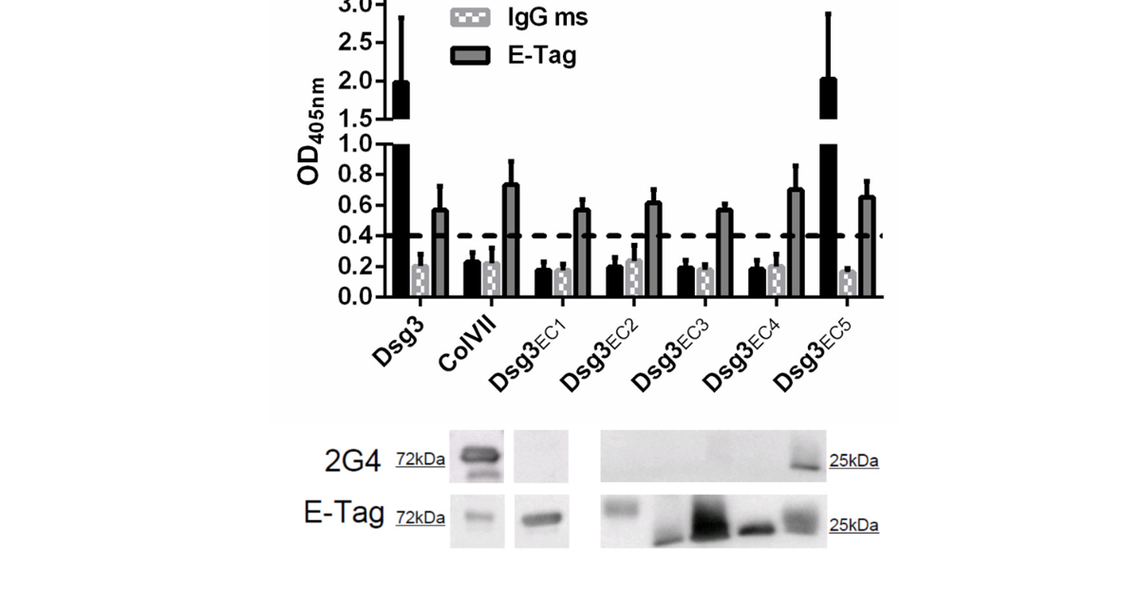
Pemphigus vulgaris (PV) is a severe autoimmune blistering disease characterized by IgG autoantibodies (auto-ab) against the desmosomal adhesion molecules desmoglein 3 (Dsg3) and Dsg1. Underlying mechanisms leading to blister formation upon binding of Dsg-specific IgG auto-ab are not fully understood. Numerous studies demonstrated the pathogenicity of IgG auto-ab binding to the amino-terminal region 1 (EC1) of the Dsg3 ectodomain. However, auto-ab in PV are polyclonal, including IgG against both aminoterminal- and membrane-proximal epitopes of the Dsg3 ectodomain.
In this study, the pathogenicity of a previously uncharacterized murine monoclonal IgG antibody, 2G4, directed against the membrane-proximal region (EC5) of the Dsg3 ectodomain, was characterized and tested in various specificity and functionality assays. The results clearly demonstrate that 2G4 is capable of inhibiting intercellular keratinocyte adhesion and of inducing cellular Dsg3 redistribution by activation of the p38MAPK signal transduction pathway. Here, we provide evidence that an IgG auto-ab directed against the membrane-proximal region EC5 of Dsg3 induces acantholysis, the hallmark in PV. These findings challenge the current concept that IgG auto-ab targeting the NH2-terminal portion of the Dsg3 ectodomain are pathogenic only. Our study provides further aspects for a deeper understanding of desmosomal keratinocyte adhesion and improves our insight on the complex auto-ab induced blister formation in PV.
Contact
Christoph Hudemann, PhD
Tel.: (+49) 06421 - 58 64823
Mail: hudemanc@staff.uni-marburg.de
Department of Dermatology and Allergology
Philipps-Universität Marburg
Baldingerstraße
D-35043 Marburg